Why we create Cardio-SM
The clinical interpretation of genetic variants in hypertrophic cardiomyopathy (HCM) is particularly challenging due to the marked heterogeneity of the condition, in terms of clinical presentation as well as its genetics.
This complexity can be managed using a rigorous, standardized, and disease/gene-specific framework for variant classification, ideally aligned with the latest ACMG/AMP recommendations as refined by variant curation expert panels (VCEP).
However, the collection and evaluation of multiple data from different sources are needed to apply this framework properly, often causing confusion during the process.
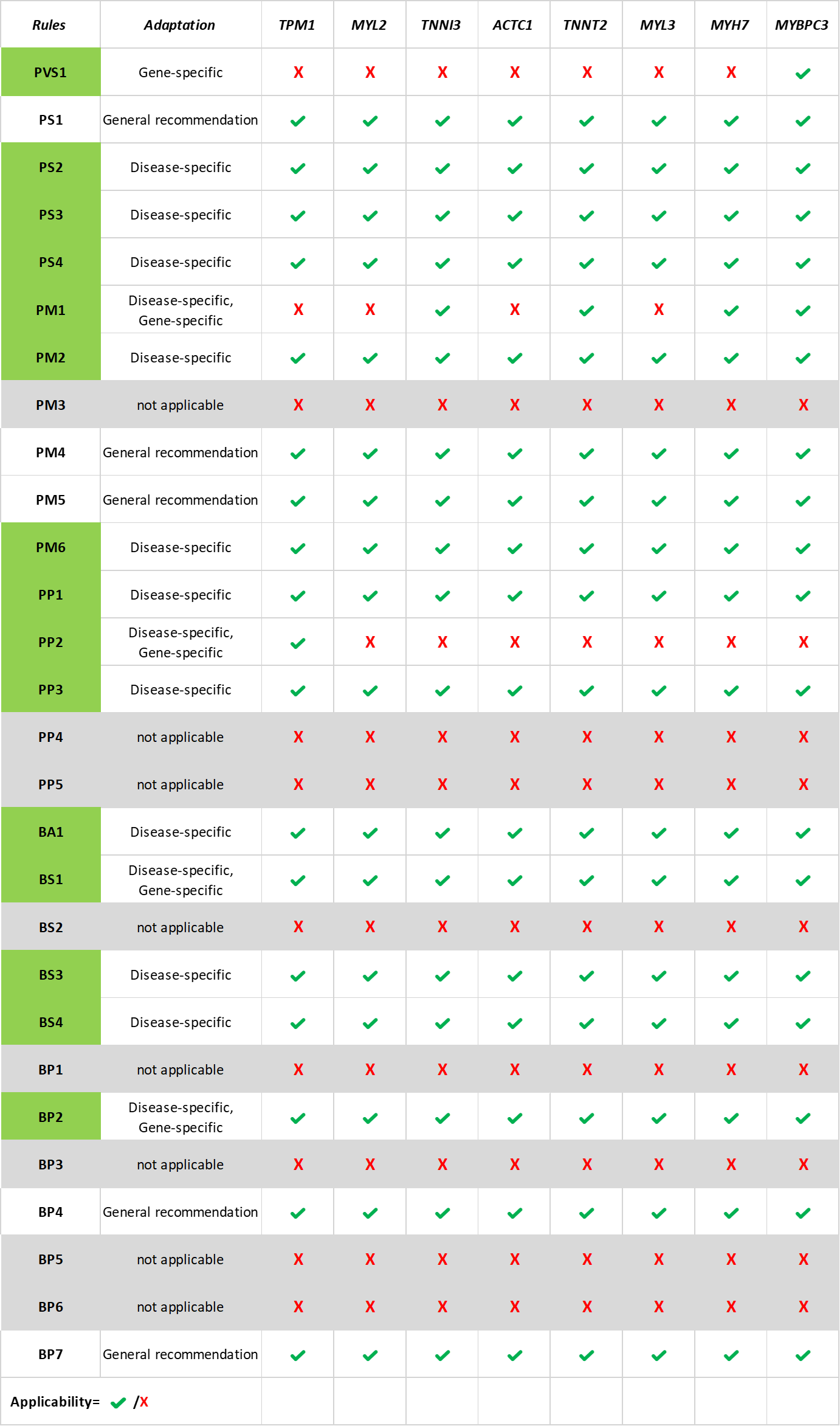
To meet this need, we developed Cardio‑SM, a semi-automated variant classification tool that enables the structured application of ClinGen Cardiomyopathy VCEP specifications for eight sarcomeric HCM genes.
The platform is intended to assist medical geneticists, laboratory professionals, and variant curators in achieving consistent, reproducible, and evidence-based interpretations in cardiovascular genetics.
Variant interpretation
We grouped the criteria into nine evidence topics (population frequency (PM2, BA1, BS1), variant type (PVS1, PM4, PP2, BP7), in silico prediction (PP3, BP4), prevalence (PS4), mutational hotspots (PM1, PS1, PM5), functional effect (PS3, BS3), allelic origin (PS2, PM6), segregation (PP1, BS4), and co-occurrence (BP2)) where each one triggers the corresponding criteria based on ClinGen Cardiomyopathy VCEP specifications.
Our interactive interface guide curators through variant classification process by prompting them to enter relevant data or to answer conceptual questions aligned with the ACMG/AMP framework. Based on the selected evidence, Cardio‑SM provides a final classification both under the standard combination rules and the quantitative point-based system along with a summary of applied rules for downstream validation.
The tool is developed in Python and hosted on GitHub